- Data Sources
- Graph Schema
- Getting Started
- Examples
- Access
- Traverse the Graph
- GitHub
- Chat
Explore
Analyze
Collaborate
Graph Info
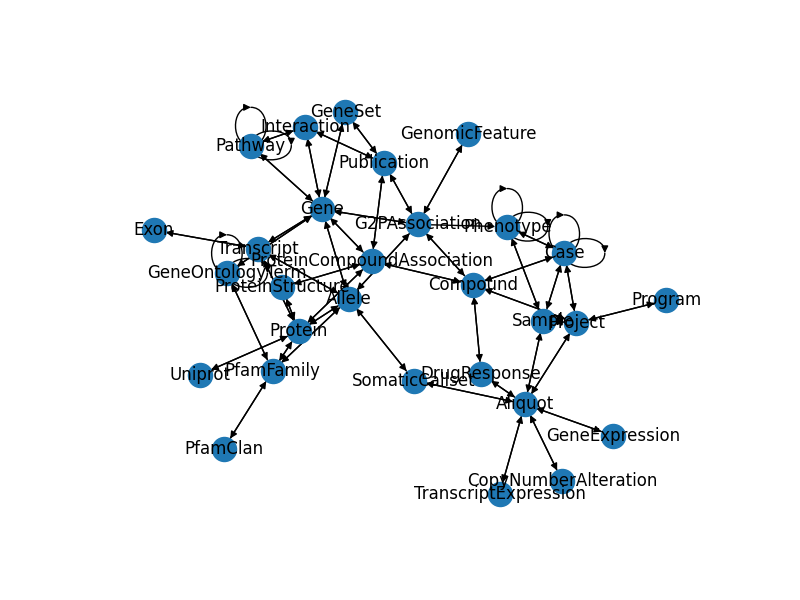
The GripQL API allows a user to download the schema of a graph. This outlines the different types of nodes, the edges the connect them and the structure of the documents stored in graph elements. A graph document has a graph field that has the name, a vertices field and an edges field.
{
"graph": "rc5",
"vertices": [
{"_id": "Compound",
"_label": "Compound",
"name": "STRING",
"term": "STRING",
"term_id": "STRING"
},
...],
"edges": [
{"_id": "(Project)--program->(Program)",
"_label": "program",
"_from": "Project",
"_to": "Program",
}
...]
}
Connect to BMEG server
import networkx as nx
import matplotlib.pyplot as plt
import gripql
from networkx.drawing.nx_agraph import graphviz_layout
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
Print avalible graphs
print(conn.listGraphs())
[‘rc6’, ‘rc6__schema__’, ‘rc6_1’, ‘rc6_1__schema__’, ‘rc5’]
Get the schema graph
schema = conn.getSchema("rc6_1")
Start build graph using NetworkX
g = nx.MultiDiGraph()
for v in schema['vertices']:
g.add_node(v['_id'])
for e in schema['edges']:
g.add_edge(e['_from'], e['_to'])
Draw Schema Graph
pos = graphviz_layout(g, prog='neato', args='')
fig, ax = plt.subplots(1, 1, figsize=(8, 6));
nx.draw(g, pos, ax=ax, with_labels=True)
plt.show()
Allele Lookup
import hashlib
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
BMEG stores alleles using a hashed version of the alteration description
genome:chromosome:start:reference_bases:alternate_bases
So an example allele would be:
GRCh37:1:27100988:C:T
Which is then run though a sha1 hash to get the string
0b0a7a23d57414e768677a6cbd764563922209df
def allele_gid(genome: str, chromosome: str, start: int,
reference_bases: str, alternate_bases: str):
if not all(v is not None for v in [genome, chromosome, start,
reference_bases, alternate_bases]):
raise ValueError("one or more args was None")
start = int(start)
if reference_bases == "-" or alternate_bases == "-":
pass
elif reference_bases[0] != alternate_bases[0]:
pass
elif len(reference_bases) > len(alternate_bases):
common = os.path.commonprefix([reference_bases, alternate_bases])
reference_bases = reference_bases[len(common):]
if len(alternate_bases) == len(common):
alternate_bases = "-"
else:
alternate_bases = alternate_bases[len(common):]
start += len(common)
elif len(reference_bases) < len(alternate_bases):
common = os.path.commonprefix([reference_bases, alternate_bases])
alternate_bases = alternate_bases[len(common):]
if len(reference_bases) == len(common):
reference_bases = "-"
else:
reference_bases = reference_bases[len(common):]
vid = "{}:{}:{}:{}:{}".format(genome, chromosome, start, reference_bases, alternate_bases)
vid = vid.encode('utf-8')
vidhash = hashlib.sha1()
vidhash.update(vid)
vidhash = vidhash.hexdigest()
return "Allele:{}".format(vidhash)
chrom = 1
loc = 27100988
ids = []
for r in ['A', 'C', 'G', 'T']:
for a in ['A', 'C', 'G', 'T']:
ids.append( allele_gid("GRCh37", chrom, loc, r, a) )
for row in G.V(ids):
print( row )
{’_id’: ‘Allele:a457a505c1551c4dbd936cddb388b021422bc0bc’, ‘_label’: ‘Allele’, ‘all_effects’: ‘ARID1A,stop_gained,p.Gln1424Ter,ENST00000324856,NM_006015.4;ARID1A,stop_gained,p.Gln1041Ter,ENST00000374152,;ARID1A,stop_gained,p.Gln321Ter,ENST00000430799,;ARID1A,intron_variant,,ENST00000457599,NM_139135.2;ARID1A,intron_variant,,ENST00000540690,;ARID1A,intron_variant,,ENST00000466382,;ARID1A,upstream_gene_variant,,ENST00000532781,;’, ‘alternate_bases’: ‘T’, ‘amino_acids’: ‘Q/’, ‘biotype’: ‘protein_coding’, ‘cdna_position’: ‘4641/8577’, ‘cds_position’: ‘4270/6858’, ‘chromosome’: ‘1’, ‘codons’: ‘Cag/Tag’, ’end’: 27100988, ’ensembl_gene’: ‘ENSG00000117713’, ’ensembl_protein’: ‘ENSP00000320485’, ’ensembl_transcript’: ‘ENST00000324856’, ’exon_number’: ‘18/20’, ‘genome’: ‘GRCh37’, ‘hgnc_id’: ‘11110’, ‘hgvsc’: ‘c.4270C>T’, ‘hgvsp’: ‘p.Gln1424Ter’, ‘hgvsp_short’: ‘p.Q1424’, ‘hugo_symbol’: ‘ARID1A’, ‘impact’: ‘HIGH’, ‘project_id’: ‘Project:Reference’, ‘protein_position’: ‘1424/2285’, ‘reference_bases’: ‘C’, ‘start’: 27100988, ‘strand’: ‘+’, ‘submitter_id’: None, ‘variant_classification’: ‘Nonsense_Mutation’, ‘variant_type’: ‘SNP’}
Gene Info
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
Look up a gene by its hugo symbol
gids = G.V().hasLabel("Gene").has(gripql.eq("$.symbol", "BRCA1")).render("_id").execute()
[INFO] 2020-01-14 14:11:07,060 1 results received in 0 seconds
Find some of the Gene Ontology terms the gene is linked to
for ent in G.V(gids).out("gene_ontology_terms").limit(10):
print(ent["_id"], ent["definition"])
[INFO] 2020-01-14 14:11:09,590 10 results received in 0 seconds
GeneOntologyTerm:GO:0000724 The error-free repair of a double-strand break in DNA in which the broken DNA molecule is repaired using homologous sequences. A strand in the broken DNA searches for a homologous region in an intact chromosome to serve as the template for DNA synthesis. The restoration of two intact DNA molecules results in the exchange, reciprocal or nonreciprocal, of genetic material between the intact DNA molecule and the broken DNA molecule.
GeneOntologyTerm:GO:0000800 A proteinaceous core found between sister chromatids during meiotic prophase.
GeneOntologyTerm:GO:0003713 A protein or a member of a complex that interacts specifically and non-covalently with a DNA-bound DNA-binding transcription factor to activate the transcription of specific genes. Coactivators often act by altering chromatin structure and modifications. For example, one class of transcription coregulators modifies chromatin structure through covalent modification of histones. A second ATP-dependent class modifies the conformation of chromatin. Another type of coregulator activity is the bridging of a DNA-binding transcription factor to the basal transcription machinery. The Mediator complex, which bridges transcription factors and RNA polymerase, is also a transcription coactivator.
GeneOntologyTerm:GO:0003723 Interacting selectively and non-covalently with an RNA molecule or a portion thereof.
GeneOntologyTerm:GO:0004842 Catalysis of the transfer of ubiquitin from one protein to another via the reaction X-Ub + Y --> Y-Ub + X, where both X-Ub and Y-Ub are covalent linkages.
GeneOntologyTerm:GO:0005515 Interacting selectively and non-covalently with any protein or protein complex (a complex of two or more proteins that may include other nonprotein molecules).
GeneOntologyTerm:GO:0005634 A membrane-bounded organelle of eukaryotic cells in which chromosomes are housed and replicated. In most cells, the nucleus contains all of the cell's chromosomes except the organellar chromosomes, and is the site of RNA synthesis and processing. In some species, or in specialized cell types, RNA metabolism or DNA replication may be absent.
GeneOntologyTerm:GO:0005654 That part of the nuclear content other than the chromosomes or the nucleolus.
GeneOntologyTerm:GO:0005737 All of the contents of a cell excluding the plasma membrane and nucleus, but including other subcellular structures.
GeneOntologyTerm:GO:0006302 The repair of double-strand breaks in DNA via homologous and nonhomologous mechanisms to reform a continuous DNA helix.
Sample Counts
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
Count number of Projects per Program
q = G.V().hasLabel("Program").as_("p").out("projects").select("p")
q = q.aggregate(gripql.term("project_count", "$._id"))
for row in q.execute()[0]["project_count"]["buckets"]:
print("%s\t%s" % (row["key"], row["value"]))
[INFO] 2020-01-14 14:25:03,363 1 results received in 0 seconds
Program:TCGA 33
Program:GTEx 31
Program:CCLE 1
Program:GDSC 1
Program:CTRP 1
Count number of Cases per Program
q = G.V().hasLabel("Program").as_("p").out("projects").out("cases").select("p")
q = q.aggregate(gripql.term("sample_count", "$._id"))
for row in q.execute()[0]["sample_count"]["buckets"]:
print("%s\t%s" % (row["key"], row["value"]))
Program:TCGA 11315 Program:GTEx 8859 Program:CCLE 1623 Program:PRISM 568
Count number of Samples per Program
q = G.V().hasLabel("Program").as_("p").out("projects").out("cases").out("samples").select("p")
q = q.aggregate(gripql.term("sample_count", "$._id"))
for row in q.execute()[0]["sample_count"]["buckets"]:
print("%s\t%s" % (row["key"], row["value"]))
Program:GTEx 204512 Program:TCGA 33405 Program:CCLE 1623 Program:PRISM 568
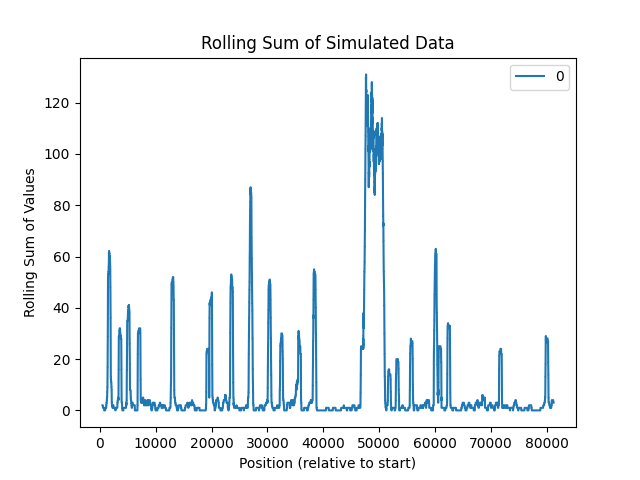
Gene Mutation Hotstops
For this example, we will start from a single gene, and identify all mutations that occur on it.
import matplotlib.pyplot as plt
import pandas
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
Get BRCA1 start and stop locations
loc = G.V().hasLabel("Gene").has(gripql.eq("symbol", "BRCA1")).render(["$.start", "$.end"]).execute()[0]
[INFO] 2020-01-14 14:12:16,554 1 results received in 0 seconds
Run an aggregation query to count up all the mutations
counts = [0] * (loc[1]-loc[0])
q = G.V().hasLabel("Gene").has(gripql.eq("symbol", "BRCA1"))
q = q.out("alleles").has(gripql.and_(gripql.eq("variant_type", "SNP"),
gripql.gte("start", loc[0]),
gripql.lte("end", loc[1])))
for v in q.aggregate(gripql.term("brac1_pos", "start")):
counts[ v['key'] - loc[0] ] = v['value']
[INFO] 2020-01-14 14:17:12,740 1 results received in 0 seconds
Save as a dataframe
s = pandas.DataFrame(counts)
Plot the hotspots
rolling_sum = s.rolling(500).sum()
rolling_sum.plot(title="Rolling Sum of Simulated Data")
plt.xlabel("Position (relative to start)")
plt.ylabel("Rolling Sum of Values")
plt.show()
<matplotlib.axes._subplots.AxesSubplot at 0x122303e10>
Cohort Genomics
Connect to BMEG server
import matplotlib.pyplot as plt
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
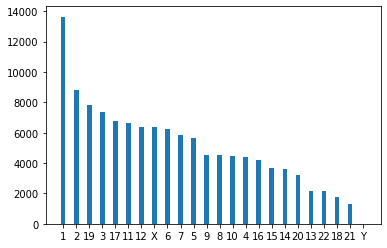
Do a query that starts on the TCGA BRCA cohort, goes though Cases -> Samples -> Aliquots -> SomaticCallsets -> Alleles. Once at the alleles, do an aggrigation to count the number of times each chromsome occurs
q = G.V("Project:TCGA-BRCA").out("cases").out("samples")
q = q.has(gripql.eq("gdc_attributes.sample_type", "Primary Tumor"))
q = q.out("aliquots").out("somatic_callsets").out("alleles")
q = q.has(gripql.eq("variant_type", "SNP"))
q = q.aggregate(gripql.term("chrom", "chromosome"))
res = q.execute()
[INFO] 2020-01-14 14:24:22,453 1 results received in 9 seconds
Visualize the results
name = []
count = []
for i in res[0].chrom.buckets:
name.append(i["key"])
count.append(i["value"])
plt.bar(name, count, width=0.35)
<BarContainer object of 24 artists>
Expression Data
import seaborn as sns
import pandas
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
Download gene expression values from TCGA-READ cohort and build matrix with submitter id as label
c = G.V("Project:TCGA-READ").out("cases").out("samples").as_("sample")
c = c.out("aliquots").out("gene_expressions").as_("exp")
c = c.render( ["$sample.gdc_attributes.submitter_id", "$exp.values"])
data = {}
for row in c.execute(stream=True):
data[row[0]] = row[1]
[INFO] 2020-01-14 14:06:01,252 177 results received in 35 seconds
Take the data we downloaded and turn it into a Pandas data frame
samples = pandas.DataFrame(data).transpose().fillna(0.0)
Take a look at the top corner of the dataframe
samples.iloc[:5,:5]
| ENSG00000000003 | ENSG00000000005 | ENSG00000000419 | ENSG00000000457 | ENSG00000000460 | |
|---|---|---|---|---|---|
| TCGA-G5-6233-01A | 30.212803 | 0.063489 | 87.317567 | 6.237288 | 5.518520 |
| TCGA-AG-4021-01A | 80.356975 | 3.621759 | 48.649980 | 8.770929 | 8.975365 |
| TCGA-EI-6514-01A | 142.160017 | 2.460405 | 82.308962 | 4.144567 | 3.251904 |
| TCGA-AG-3725-01A | 81.611032 | 4.730710 | 56.783610 | 4.896992 | 4.208633 |
| TCGA-AG-3725-11A | 78.390851 | 1.037580 | 51.193885 | 6.455982 | 2.212233 |
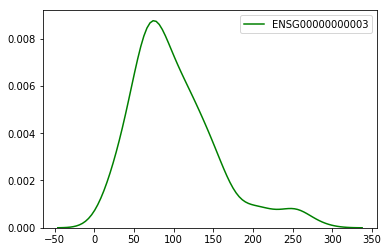
Take a quick look to see the top expressing samples for the gene ENSG00000000003
samples["ENSG00000000003"].sort_values(ascending=False).head()
TCGA-DC-5869-01A 281.005900
TCGA-DC-6683-01A 258.010380
TCGA-AF-3913-01A 254.599210
TCGA-EF-5831-01A 253.774315
TCGA-DC-6157-01A 251.170374
Name: ENSG00000000003, dtype: float64
sns.kdeplot(samples['ENSG00000000003'], color="g")
<matplotlib.axes._subplots.AxesSubplot at 0x11ceef450>
Differential Expression
import matplotlib.pyplot as plt
import seaborn as sns
import pandas
from scipy.stats import ttest_ind
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
Look at the expression in the TCGA LUAD project
PROJECT="Project:TCGA-LUAD"
Starting from the TCGA-LUAD project, follow the edges from Project -> Case -> Sample -> Aliquot -> GeneExpression. At the Sample node, we select from Solid Tissue Normal so we only pull the normals during this query.
Once on the gene expression node, extract the values, which hold the gene expression values in a Map[String,Float] format with Ensembl Gene ids as the keys, and the Gene expression TPMs as the values. We then load that into a Pandas data frame, and transpose so that the rows are the sample ids.
c = G.V(PROJECT).out("cases").out("samples").as_("sample")
c = c.has(gripql.eq("gdc_attributes.sample_type", "Solid Tissue Normal"))
c = c.out("aliquots").out("gene_expressions").as_("exp")
c = c.render( ["$sample.gdc_attributes.submitter_id", "$exp.values"])
data = {}
for row in c.execute(stream=True):
data[row[0]] = row[1]
normalDF = pandas.DataFrame(data).transpose()
[INFO] 2020-01-14 14:07:38,218 59 results received in 12 seconds
Do the Project to Gene Expression traversal again, but this time only select the tumor samples.
c = G.V(PROJECT).out("cases").out("samples").as_("sample")
c = c.has(gripql.eq("gdc_attributes.sample_type", "Primary Tumor"))
c = c.out("aliquots").out("gene_expressions").as_("exp")
c = c.render( ["$sample.gdc_attributes.submitter_id", "$exp.values"])
data = {}
for row in c.execute(stream=True):
data[row[0]] = row[1]
tumorDF = pandas.DataFrame(data).transpose()
[INFO] 2020-01-14 14:09:27,596 539 results received in 107 seconds
For each gene, run t-test to determine the genes with the most differential expression between the tumor and normal sets.
stats = {}
for gene in tumorDF:
s = ttest_ind(tumorDF[gene], normalDF[gene])
stats[gene] = { 'statistic': s.statistic, 'pvalue' : s.pvalue }
statsDF = pandas.DataFrame(stats).transpose()
Display the results
statsDF[ statsDF['pvalue'] < 0.0001 ].sort_values('statistic').head()
| statistic | pvalue | |
|---|---|---|
| ENSG00000168484 | -53.356952 | 6.389050e-227 |
| ENSG00000135604 | -48.643308 | 9.675744e-208 |
| ENSG00000204305 | -46.262534 | 1.283753e-197 |
| ENSG00000114854 | -44.158458 | 2.022993e-188 |
| ENSG00000022267 | -40.822561 | 2.395204e-173 |
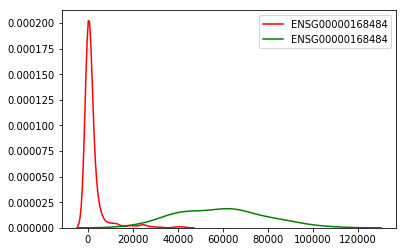
Using the top gene from the T-Test experiment (ENSG00000168484), plot the expression of the gene across the tumor and normal samples
sns.kdeplot(tumorDF['ENSG00000168484'], color="r")
sns.kdeplot(normalDF['ENSG00000168484'], color="g")
<matplotlib.axes._subplots.AxesSubplot at 0x11677b8d0>
Do a quick search of ENSG00000168484 to identify the Gene Ontology terms that it is linked to
for row in G.V("ENSG00000168484").out("gene_ontology_terms"):
print(row._id, row.definition)
[INFO] 2020-01-14 14:10:14,516 10 results received in 0 seconds
GeneOntologyTerm:GO:0005515 Interacting selectively and non-covalently with any protein or protein complex (a complex of two or more proteins that may include other nonprotein molecules).
GeneOntologyTerm:GO:0005576 The space external to the outermost structure of a cell. For cells without external protective or external encapsulating structures this refers to space outside of the plasma membrane. This term covers the host cell environment outside an intracellular parasite.
GeneOntologyTerm:GO:0005615 That part of a multicellular organism outside the cells proper, usually taken to be outside the plasma membranes, and occupied by fluid.
GeneOntologyTerm:GO:0005789 The lipid bilayer surrounding the endoplasmic reticulum.
GeneOntologyTerm:GO:0007585 The process of gaseous exchange between an organism and its environment. In plants, microorganisms, and many small animals, air or water makes direct contact with the organism's cells or tissue fluids, and the processes of diffusion supply the organism with dioxygen (O2) and remove carbon dioxide (CO2). In larger animals the efficiency of gaseous exchange is improved by specialized respiratory organs, such as lungs and gills, which are ventilated by breathing mechanisms.
GeneOntologyTerm:GO:0042599 A membrane-bounded organelle, specialized for the storage and secretion of various substances (surfactant phospholipids, glycoproteins and acid phosphates) which are arranged in the form of tightly packed, concentric, membrane sheets or lamellae. Has some similar properties to, but is distinct from, a lysosome.
GeneOntologyTerm:GO:0042802 Interacting selectively and non-covalently with an identical protein or proteins.
GeneOntologyTerm:GO:0044267 The chemical reactions and pathways involving a specific protein, rather than of proteins in general, occurring at the level of an individual cell. Includes cellular protein modification.
GeneOntologyTerm:GO:0045334 A clathrin-coated, membrane-bounded intracellular vesicle formed by invagination of the plasma membrane around an extracellular substance.
GeneOntologyTerm:GO:0097486 The volume enclosed by the outermost membrane of a multivesicular body.
CNA Histogram
import matplotlib.pyplot as plt
import numpy as np
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
Get Ensembl Gene ids for genes of interest
GENES = ["PTEN", "TP53", "RB1"]
gene_ids = {}
for g in GENES:
for i in G.V().hasLabel("Gene").has(gripql.eq("symbol", g)):
gene_ids[g] = i["_id"]
gene_ids
{'PTEN': 'ENSG00000171862',
'TP53': 'ENSG00000141510',
'RB1': 'ENSG00000139687'}
For each gene of interest, obtain the copy number alteration values and aggregate them by gene.
q = G.V("Project:TCGA-PRAD").out("cases").out("samples").out("aliquots")
q = q.has(gripql.eq("$.gdc_attributes.sample_type", 'Primary Tumor')).out("copy_number_alterations")
q = q.aggregate(
list( gripql.term( g, "values.%s" % (g), 5) for g in gene_ids.values() )
)
res = list(q)
for item in res:
print(f"{item['name']}\t{item['key']}:{item['value']}")
ENSG00000171862 0:327 ENSG00000171862 -2:95 ENSG00000171862 -1:64 ENSG00000171862 1:5 ENSG00000171862 2:1 ENSG00000141510 0:329 ENSG00000141510 -1:126 ENSG00000141510 -2:37 ENSG00000139687 0:269 ENSG00000139687 -1:139 ENSG00000139687 -2:81 ENSG00000139687 1:3
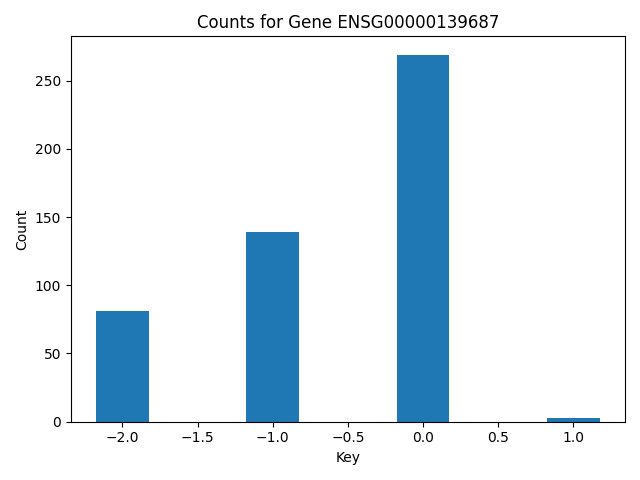
Create a barchart showing the counts of copy number altered samples in the cohort.
val = []
count = []
gene_data = [item for item in res if item['name'] == 'ENSG00000139687']
sorted_data = sorted(gene_data, key=lambda x: x['key'])
val = [item['key'] for item in sorted_data]
count = [item['value'] for item in sorted_data]
plt.bar(val, count, width=0.35)
plt.xlabel("Key")
plt.ylabel("Count")
plt.title("Counts for Gene ENSG00000139687")
plt.tight_layout()
plt.show()
Kaplan Meier
Install the lifelines library
!pip install lifelines
Collecting lifelines
[?25l Downloading https://files.pythonhosted.org/packages/a5/8e/56c7d3bba5cf2f579a664c553900a2273802e0582bd4bdd809cdd6755b01/lifelines-0.23.6-py2.py3-none-any.whl (407kB)
[K |████████████████████████████████| 409kB 4.1MB/s eta 0:00:01
[?25hCollecting autograd>=1.3
Downloading https://files.pythonhosted.org/packages/23/12/b58522dc2cbbd7ab939c7b8e5542c441c9a06a8eccb00b3ecac04a739896/autograd-1.3.tar.gz
Requirement already satisfied: scipy>=1.0 in /usr/local/lib/python3.7/site-packages (from lifelines) (1.3.1)
Collecting matplotlib>=3.0
Using cached https://files.pythonhosted.org/packages/a0/76/68bc3374ffa2d8d3dfd440fe94158fa8aa2628670fa38bdaf186c9af0d94/matplotlib-3.1.2-cp37-cp37m-macosx_10_9_x86_64.whl
Collecting autograd-gamma>=0.3
Downloading https://files.pythonhosted.org/packages/3e/87/788c4bf90cc5c534cb3b7fdb5b719175e33e2658decce75e35e2ce69766f/autograd_gamma-0.4.1-py2.py3-none-any.whl
Requirement already satisfied: numpy>=1.14.0 in /usr/local/lib/python3.7/site-packages (from lifelines) (1.17.3)
Requirement already satisfied: pandas>=0.23.0 in /usr/local/lib/python3.7/site-packages (from lifelines) (0.25.1)
Requirement already satisfied: future>=0.15.2 in /usr/local/lib/python3.7/site-packages (from autograd>=1.3->lifelines) (0.17.1)
Requirement already satisfied: pyparsing!=2.0.4,!=2.1.2,!=2.1.6,>=2.0.1 in /usr/local/lib/python3.7/site-packages (from matplotlib>=3.0->lifelines) (2.2.0)
Requirement already satisfied: python-dateutil>=2.1 in /usr/local/lib/python3.7/site-packages (from matplotlib>=3.0->lifelines) (2.7.3)
Requirement already satisfied: cycler>=0.10 in /usr/local/lib/python3.7/site-packages (from matplotlib>=3.0->lifelines) (0.10.0)
Requirement already satisfied: kiwisolver>=1.0.1 in /usr/local/lib/python3.7/site-packages (from matplotlib>=3.0->lifelines) (1.0.1)
Requirement already satisfied: pytz>=2017.2 in /usr/local/lib/python3.7/site-packages (from pandas>=0.23.0->lifelines) (2018.5)
Requirement already satisfied: six>=1.5 in /usr/local/lib/python3.7/site-packages (from python-dateutil>=2.1->matplotlib>=3.0->lifelines) (1.12.0)
Requirement already satisfied: setuptools in /usr/local/lib/python3.7/site-packages (from kiwisolver>=1.0.1->matplotlib>=3.0->lifelines) (42.0.2)
Building wheels for collected packages: autograd
Building wheel for autograd (setup.py) ... [?25ldone
[?25h Created wheel for autograd: filename=autograd-1.3-cp37-none-any.whl size=47990 sha256=a14ecb3ba1b6a4626c2721a672f67abab2d2726c463406a31951fcff0effe5d6
Stored in directory: /Users/strucka/Library/Caches/pip/wheels/42/62/66/1121afe23ff96af4e452e0d15e68761e3f605952ee075ca99f
Successfully built autograd
Installing collected packages: autograd, matplotlib, autograd-gamma, lifelines
Found existing installation: matplotlib 2.2.2
Uninstalling matplotlib-2.2.2:
Successfully uninstalled matplotlib-2.2.2
Successfully installed autograd-1.3 autograd-gamma-0.4.1 lifelines-0.23.6 matplotlib-3.1.2
from lifelines import KaplanMeierFitter
import matplotlib.pyplot as plt
import pandas
import gripql
conn = gripql.Connection("https://bmeg.io/api", credential_file="bmeg_credentials.json")
G = conn.graph("rc6_1")
Look at the TCGA-BRCA cohort, and find all of the cases where there is a recorded days_to_death
q = G.V("Project:TCGA-BRCA").out("cases")
data = {}
for i in q:
if i.gdc_attributes.demographic is not None and i.gdc_attributes.demographic.vital_status == "Dead":
if 'days_to_death' in i.gdc_attributes.demographic:
data[ i._id ] = i.gdc_attributes.demographic.days_to_death
survival = pandas.Series(data)
[INFO] 2020-01-14 14:18:49,810 1,098 results received in 0 seconds
Gene ensembl gene id for TP53
gene = G.V().hasLabel("Gene").has(gripql.eq("symbol", "TP53")).execute()[0].gene_id
print(gene)
[INFO] 2020-01-14 14:19:08,643 1 results received in 0 seconds
ENSG00000141510
Starting from the cases with attached survival information, find all of the cases that have a mutation in the gene on interest
q = G.V(list(survival.keys())).as_("case").out("samples").out("aliquots").out("somatic_callsets").out("alleles")
q = q.has(gripql.eq("ensembl_gene", gene))
q = q.select("case").distinct("$._id").render("$._id")
mut_cases = list(q)
[INFO] 2020-01-14 14:19:47,044 52 results received in 2 seconds
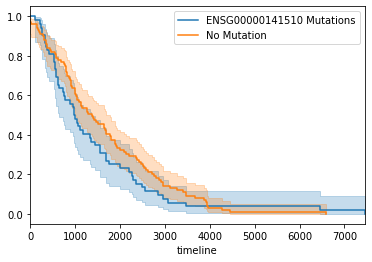
Plot a Kaplan Meirer curve to demonstrate the different in survival of the somatic mutation group and those with no somatic mutation.
kmf = KaplanMeierFitter()
ax = plt.subplot(111)
kmf.fit(survival[mut_cases], label="%s Mutations" % (gene))
ax = kmf.plot(ax=ax)
kmf.fit(survival[ survival.index.difference(mut_cases) ], label="No Mutation")
kmf.plot(ax=ax)
<matplotlib.axes._subplots.AxesSubplot at 0x1205a8d10>